Science
Bioavailability of CBD
R&D is essential to deliver on our purpose of creating world-changing products for a better every day. CS MEDICA combines research, technology, and nature within healthcare to revolutionize with innovative treatments and technology within the cannabis and medical industries.
Tapping into unexploited potential
To ensure that we deliver value to society, R&D continuously pursues even higher levels of innovation across more therapy areas and technology platforms and with more patients and partners. In 2016, we found untapped potential in substances contained in Cannabis sativa L. that were not exploited in the treatment sector. A big part of the neglected potential was caused by a lack of confidence in the existing CBD products’ effectiveness and safety. Through extensive research leading to greater knowledge on the differences between CBD (cannabidiol), THC tetrahydrocannabinol), and other cannabinoids, the demand for products containing the substance and their respective prosperities has increased dramatically over the last years.
Working on multiple development stages
As a MedTech, we work at all Development Stages, where III, IV & V are our focus (with 23 products in scope, 9 launched):
New Product Development and Technologies.
- Product Developing, verifying, and validating, securing patents.
- Optimizing processes and trials, moving from MDD to MDR.
- Launched finished products for brand build, post-market assessment, and revenue stream.
The discovery and development of our medical device treatments are time-consuming, expensive in clinical trials, and dependent on the latest regulatory changes. To compensate for the long time to market, our R&D strives to advance our existing products by discovering potential new segments, indications, and reformulations into cosmetics, still science-backed and safe.
Our R&D strategy
- Deliver a pipeline of innovative medical devices with cannabidiol to help bring change and relief to patients’ lives worldwide with the therapeutic values of cannabinoids.
- Source the best ingredients and partnerships for safe, efficient, and accessible results for those that need a better every day.
- Advance our capabilities to position us for long-term R&D and trusted CBD leadership.
- While a considerable amount of our R&D is internal, we strive for know-how and innovative technologies developed by others to integrate into our discovery and development processes or products. We collaborate with universities, companies, and other partners, allowing us to share knowledge, and optimize operations.
Behind the science
The endocannabinoid system (ECS) is a biological system composed of endocannabinoids, which are endogenous lipid-based retrograde neurotransmitters that bind to cannabinoid receptors (CBRs), and cannabinoid receptor proteins that are expressed throughout the vertebrate central nervous system (including the brain) and peripheral nervous system. Endocannabinoids have important effects on immune functions as well. They modulate T- and B-lymphocytes proliferation and apoptosis, macrophage-mediated killing of sensitized cells, inflammatory cytokine production, immune cell activation by inflammatory stimuli, chemotaxis, and inflammatory cell migration.
The ECS under preliminary research but may be involved in regulating physiological and cognitive processes, including fertility, pregnancy, pre-and postnatal development, various activity of immune system, appetite, pain-sensation, mood, and memory, and in mediating the pharmacological effects of cannabis. The ECS plays an important role in multiple aspects of neural functions, including the control of movement and motor coordination, learning and memory, emotion and motivation, addictive-like behaviour, and pain modulation, among others. Two primary cannabinoid receptors have been identified: CB1, first cloned (or isolated) in 1990; and CB2, cloned in 1993.
Our DNA
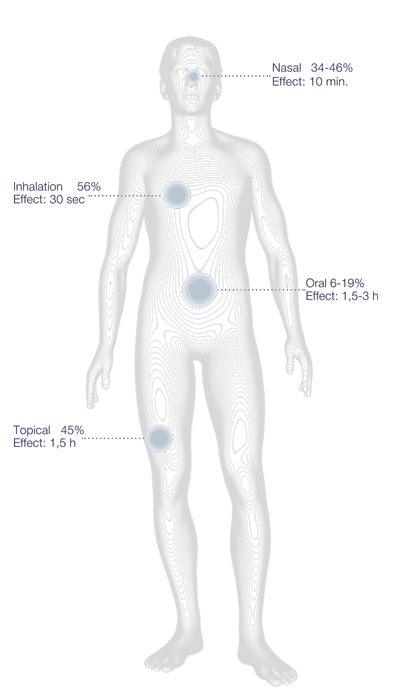
We have researched the bioavailability of the Phyto cannabinoids in the human body together with drug delivery systems to deliver cannabinoids to the human body with high efficacy. We found the oral delivery of the Phyto cannabinoids was highly ineffective, as the bioavailability was only 6% and the orally delivered Phyto cannabinoids went through the liver, where it reduces the enzyme degrading process of medicine, meaning the medicine has a higher efficacy in the body. Whereas the delivery of Phyto cannabinoids through the skin or the nasal passage has a bioavailability of 45% and does not enter the lever, as it does not enter the bloodstream. CBD reach the receptors of pain, psoriasis, hair, inflammation (wounds) and arthritis through the skin. Many of the receptors are present in the skin. CBD applied topically or nasal targets the localized area/problem, dispersing across the skin reaching muscles and cells – very little CBD enters the bloodstream and thereby does not reach the liver.

Why Natural CBD
In our research and development, we also found that several factors impact the efficacy/bioactivity of the extracted Phyto cannabinoids – why some Phyto cannabinoids extracted from Cannabis Sativa L. plant have no effect/no bioactivity in the human body, as the extracted Phyto cannabinoids must be recognizable by the human body to be utilized. An external journal1 describe that synthetic CBD have no effect /no bioactivity in the human body, as the human body do not recognize the synthetic CBD as an alternative to the body’s own produced endocannabinoid (AEA, 2-AG) – like the human body cannot recognize and utilize synthetic insulin, as the body do not recognize the synthetic insulin as the human insulin the body produces itself. If the body do not recognize the insulin – the insulin will not be able to interact with the insulin receptor and open the glucose channel in the cell.
New Product Development
Clinical Trials
Clinical trials are essential in establishing the safety and efficacy of new medical devices, including those utilizing innovative compounds like CBD. These trials provide critical data that distinguish these devices from other products in the market, ensuring they meet rigorous health standards before reaching consumers. For medical devices enhanced with CBD, known for its potential therapeutic benefits, clinical trials help validate claims about their health benefits and functionality. By participating in these structured evaluations, manufacturers can demonstrate that their products are effective and safe for consumer use. As a result, clinical trials are fundamental in elevating the credibility of CBD-enhanced medical devices, setting them apart in a crowded marketplace and advancing the frontiers of medical technology.
The Clinical Trials & Pre-Clinical studies for medical devices.
Registration ahead of competition
- The Clinical Trials & Pre-Clinical studies for medical devices. Registration ahead of competition.
- Pre-clinical trials & Biocom test finalized.
- Post-clinical trials are performed.
- Registered as one of the only OTC Medical Devices with CBD under MDD, for our treatment areas.
- Signed contract with BSI, Notify Body as the first company for Rule 21, transferring to MDR according to plan.
- 2 registered MDR treatment.
Legal & Regulatory
In the world, the definition of a medical device can vary depending on the country or region. However, a medical device is any instrument, implant, topicals or other similar or related article that is used in the diagnosis, treatment, or prevention of disease or other medical conditions. The regulation of medical devices also varies around the world, with different countries having their own regulatory agencies and laws governing the manufacturing, distribution, and use of medical devices. In the United States, for example, the Food and Drug Administration (FDA) is responsible for regulating medical devices, while in Europe the European Medicines Agency (EMA) is responsible. Other countries may have different regulatory bodies and laws, like the UK that due to Brexit has its own regulatory framework for medical devices, and the MHRA will be the competent authority for medical devices in the UK.
Before placing a medical device on the European market, technical documentation for the current class of medical devices are produced providing evidence of conformity with the relevant legislation. All technical documentation comply with the Medical Devices Directive (MDD) 93/42/EEC and in process of updating according to MDR.
Legal over-the-counter (OTC) cannabinoid products only include Cosmetics and Medical Devices delivered Topically and Intranasally. The European Medicines Agency EMA, the UK and Hong Kong have initiated a withdrawal of oral CBD oils and other CBD supplements. This directly results in a large portion of the current CBD products being removed from the market leaving only authorized medical devices and cosmetics products. As all CS MEDICA products are either Topical or Intranasal, we believe that the change in the law is in our favor and we are one of few registered at MDR in Europe and MHRA in the UK, we have 160 free sales certificates outside Europe, and possess a certificate from an authorized laboratory with proof of non-THC on a 1ppb level, needed for sports people and racehorses (anti-doping) and for the Asian markets.
Our treatment products are registered as Medical Device at the Danish Medical Agency and in EUDAMED covering all EU countries.
CANNASEN® treatment products are registered medical devices with cannabidiol under the Medical Devices Directive (93/42/EEC) (MDD), and classified as class I . Up to May 26, 2021, Medical devices was regulated under MDD, but today follows the MDR (Medical Device Regulation) (EU) 2017/745 (MDR). Products filed under MDD as a class I will, with the new MDR, be lifted to a class IIa. For a transition period of 4 years, products with a MDD class I are allowed to remain on the market, provided that the extended requirements of the classification lift are initiated, at the date changing from MDD to MDR and the classification lift is completed within the transition period.
CANNASEN® products launched as a class I under MDD are allowed to stay on the market until 2024 (and up to 2028 if EU’s proposal to prolong the grace period), as a class 1 as the extended requirements of the classification lift are initiated, at the date changing from MDD to MDR. To the best of our knowledge, CS MEDICA A/S is currently first-mover on the market with products containing cannabinoids regulated under MDR. This immediately gives a competitive advantage as new products introduced to the market under MDR with cannabinoids must go through the process applicable to MDR class IIa, corresponding to an application process period of minium 2-3 years.
All CANNASEN® products comply with the EU regulation and are registered for sale in all EU countries and with >160 free sales certificates for countries outside Europe. CBD crystals used in our Topical products is extracted from the plant called Cannabis sativa L., which is in compliance with the legal strain of hemp according to the European Common Catalogue of Varieties of Agricultural Plant Species, in accordance with Article 17 of council directive 2002/53/EC of 13 June 2002, with a level of THC < 0,2%.
Sale and marketing of legal CBD products containing CBD extracted from the plant Cannabis sativa L are approved by the supreme authority within the EU, the Court of Justice of the European Union. Court declaration, of the 19th of November, 2020, can be found in here.
Currently legal CBD products include CBD delivered topically and intra-nasally. The EU and the UK has thus initiated the withdrawal of all oral CBD oils and other CBD supplements. As a result, a portion of the current CBD products is being removed from the market leaving only authorized medical devices and cosmetics products. As these two segments are the main focuses of the CANNASEN® brand, CS medica believes that the change in the law is in the company’s favor [1]
Testing and Compliance links:
› Extracts Regulation of Cannabidiol in the EU and UK
› Latest CBD laws around europe
› Regulatory status of CBD in food in europe
› CBD will be removed from shelves next year, are you prepared?
› 2019/01/19-01-14-press_release_EIHA-novelfoods
Extensive tests and control including invitro skin-sensitization test, in-vitro efficacy test, skin-irritating test, and cytotoxicity test, are performed on all treatment products produced by CS medica’s subsidiaries. All raw materials used in our products undergo a quality assurance check to ensure consistent quality. All testing instructions meet the legal requirements according to section 3.2 in Appendix II of the MDD.
As a manufacturer with of a medical device on the EU market we comply with the specific European Directives set forth by the European Commission. In our case, of importance are the Medical Device Directives (MDD): MDD 93/42/EEC and MDR 2017/745.
Our products are registered at the Danish Medical Agency and in EUDAMED (EU database for legal medical devices) as a Medical Device Manufacturer, covering all EU countries. Additionally, we make sure to contact relevant Medical Agencies before entering new markets to assure it complies with its current regulation on medical devices and local cannabinoids legislation.
Learn more about the about the process that has been followed.
Medical devices class I under MDD requirement
The Process following medical devices class I certification under MDD requirement can be summarized as follows
Bibliography for all active ingredients in treatment products must be identified and documented through:in vitro testing, in, clinical testing, and safety data. Preliminary feasibility studies are then performed to optimize the final formulation.
Test of formulation, in accordance withICH guidelines and ISO 13485 including; viscosity test, homogeneous test, microbiology test, water content, skin sensation, smell, and color.
The test of formulation is followed by an in-vitro test, skin irritation according to ISO 10993-10, skin sensation according to OECD 442E, and cytotoxicity according to ISO 10993-5: 2009.
A determination of the shelf-life, including a stability test in accordance with ICH guidelines (the International Conference on Harmonization of Technical Requirements for the Registration of Pharmaceuticals for Human Use).
A review of product documentation, packaging, and package leaflet to make sure they follow the legislation within Medical Device and are in accordance with the MDD and MEDDEV guidelines. Any other languages than English on the package needs to be translated by a certified translator for each language needed.
Submission of a technical file according to MDD and ISO 13485, which, apart from the abovementioned, should include: bibliography, tests, and a clinical evaluation of the complete product, including; regulatory affairs, literature review,/technical specifications, risk-benefit evaluation, pre-clinical studies, clinical investigation on the actual or equivalent device, post-market surveillance with an own device and at least one equivalent device (if applicable), and a full risk assessment and biological evaluation according to iso 14971 and iso 10993 respectively.
The technical file is made to document that the medical device that encounters the patient’s body is expected to perform, its intended use/function without resulting in any adverse effect to a patient.
Determination of classification based on the medical devices s risks and the regulatory controls necessary to provide a reasonable assurance of safety and effectiveness according to the rules set in MDD.
With the MDR regulation, the requirements for demonstration of product quality, safety and reliability have been further tightened and the following general conditions required under the new MDR is in the process of being implemented by CS medica.
New Medical Device Regulation in EU
The transition from MDD to MDR represents a critical regulatory shift in the European medical device market. Companies not yet part of this transition face significant risks and challenges but also have opportunities to enhance their market position by demonstrating compliance with the latest standards. Proactive planning, early engagement with Notified Bodies, and robust implementation of MDR requirements are essential for a successful transition.
We are ahead of the competition as the first to sign Rule 21 with a Notified Body in the MDR transition. Being secured to sell throughout the transition until the end of 2028 also provides a significant advantage. Our MDR status also enables us to register under the US FDA, AUS TGA, and other stringent country regulations.
With two products already MDR compliant, our substantial investments in compliance elevate our market position.
Local Registration
Our heavy investments in this careful compliance approach underpin the success and sustainability of operations in new markets with a strong positioning as a hybrid between the pharmaceutical industry and the cannabis sector.

Patents
Patents Achieved in 2023
KINGDOM OF DENMARK
Patent No. DK 181405
Patent No. DK 181329
Patent No. DK 181458
Patents Achieved in 2024
Granted to be issued on Anti-Hair Loss serum in DK & PsoriasisGel in EU.
In process
Final tests for bioactive CBD are ready, we currently await issuing a patent application.
Processing & Production
Our CMOs deliver on certified ISO13485 and MDR 2017/745.
Farming, processing and production
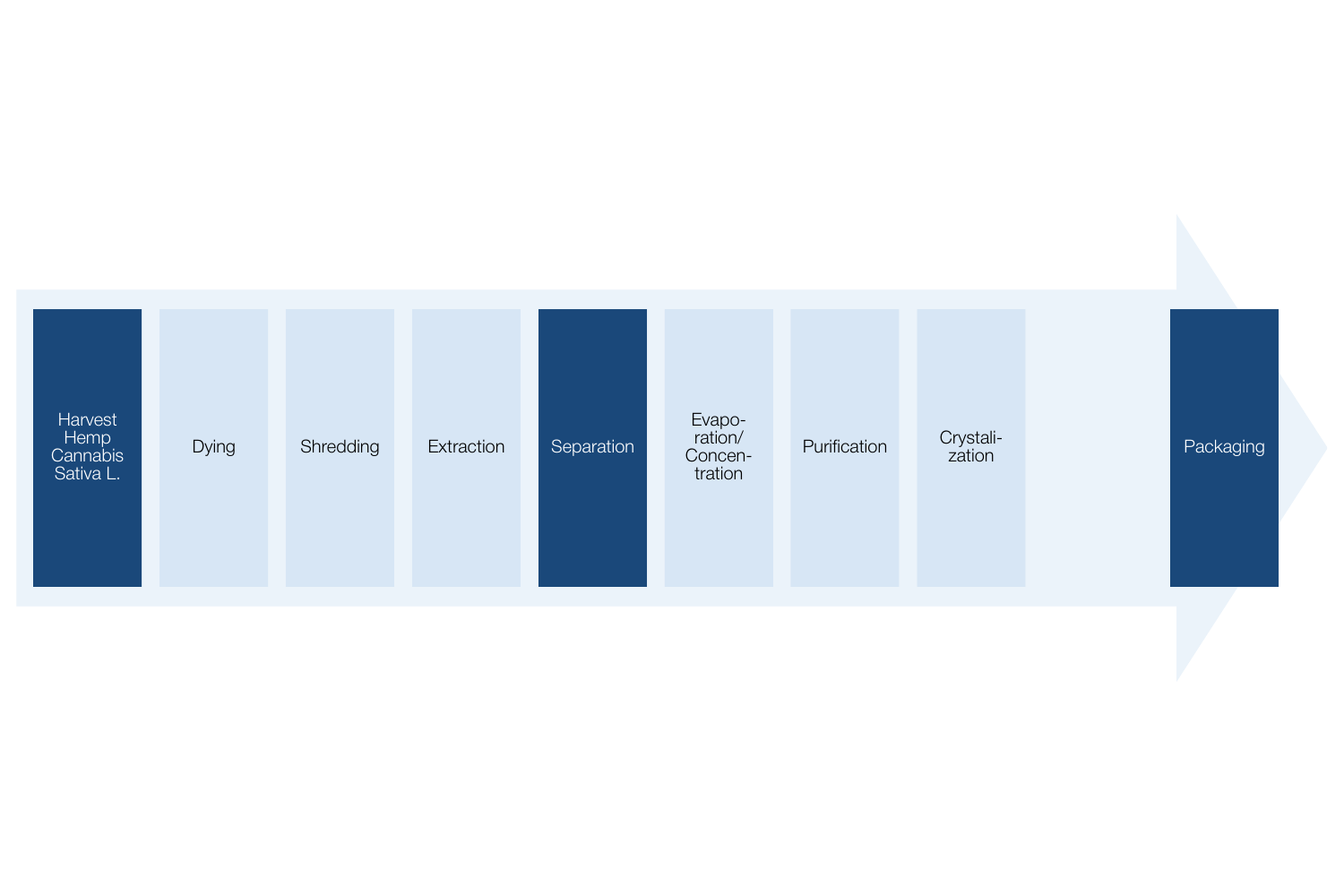
To ensure the quality of our products, we maintain control and provide transparency of our entire value chain. CBD for all our products comes from a plant called Cannabis sativa L., which is a legal strain of hemp according to the European Common Catalogue of Varieties of Agricultural Plant Species, in accordance with Article 17 of council directive 2002/53/EC of 13 June 2002.
The cultivation of Cannabis sativa L. used in our products is outsourced to Greenvalley, Italy, which specializes in industrial hemp farming. Continuous analyses are performed on our Cannabis sativa L. before the extraction process. All certificates of analysis related to cultivation are recorded and documented in our system and can be found below.
Green Valley has developed a genetic research program that allowed to identify which seeds can be specifically suitable for the production of medical devices. To minimize possible risks to health and environment, cultivation process by Greenvalley does not involve the use of pesticides or any other chemical substances. On top of that, our production follows GACP (Good Agricultural and Collection Practices).
The chemical extraction of CBD consists of separating the needed cannabinoids (i.e. CBD, CBG) from the solid plant matrix, in order to explicitly obtain rich/concentrated extracts of those components of interest, and to remove the THC. The applied extraction methods and conditions are monitored by laboratories, with special attention to process parameters including temperature, extraction agent and reagents used.
Our production process involves the use of GRAS (Class III) solvents like EtOH; and the different procedures ensure the separation of interferents (like wax) that would disturb the purification and isolation phases of a specific cannabinoid, without degrading its natural molecular structure. Such a structured process allows us to work on the acid components (i.e. CBDA) to be decarboxylated if necessary.
We evaluate every batch of product to secure that it does not contain any traceable THC, as well as that only the CBD from Cannabis sativa L. (legal strain of hemp), is used in treatment line products produced by CS MEDICA. We also ensure to contact relevant Medical Agencies before entering new markets to assure it complies with its current regulation on medical devices and local cannabinoids legislation.
In all processes, high-quality management standards in development, production, and product control are maintained according to HACCP (Hazard Analysis and Critical Control Points). All production phases follow GMP requirements (Good Manufacturing Practice, Rules – Instructions for all phases of the production cycle),and are based on a solid integrated quality system and precise risk management procedures, to ensure compliance with ICH (International Council for Harmonization – Technical Requirements for Medicinal Products for Human Use) stability guidelines, as well as ISO (International Standard Organization) quality regulations (ISO9001, ISO 10993, 14971 and ISO 13485).
Packaging
The packaging materials that come in contact with our medical devices are evaluated according to ISO 10993. Certificates that related to packaging are documented in our system and recorded in the biological evaluation report of medical device products.